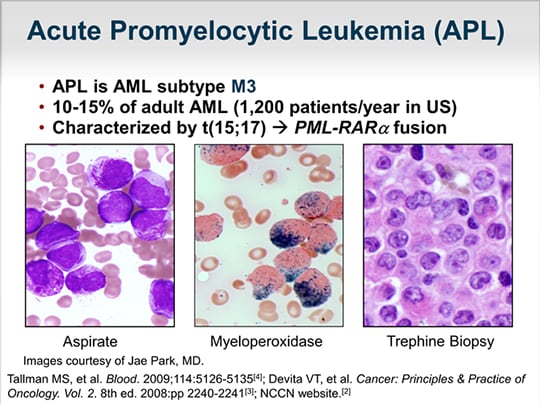
La Leucemia Promielocítica Aguda (LPA) es un subtipo agresivo de Leucemia Mieloide Aguda (LMA) caracterizada por una translocación cromosómica que involucra al gen RARα (receptor alfa del ácido retinoico).
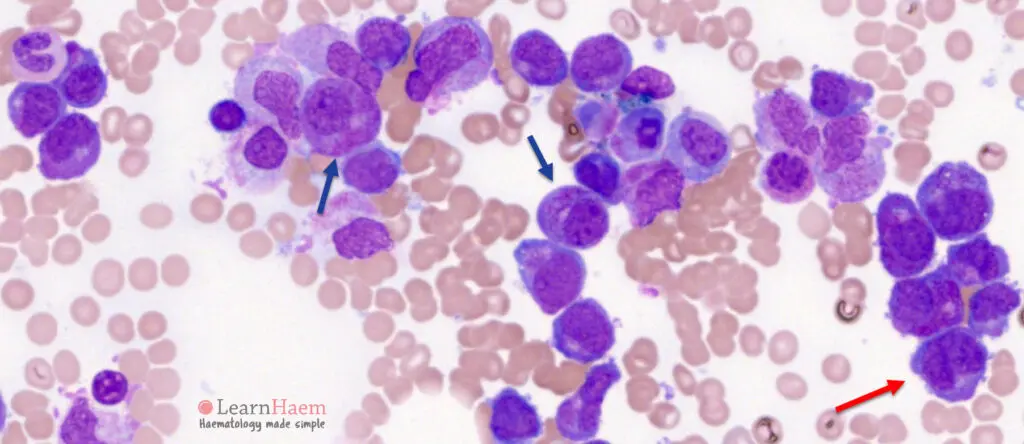
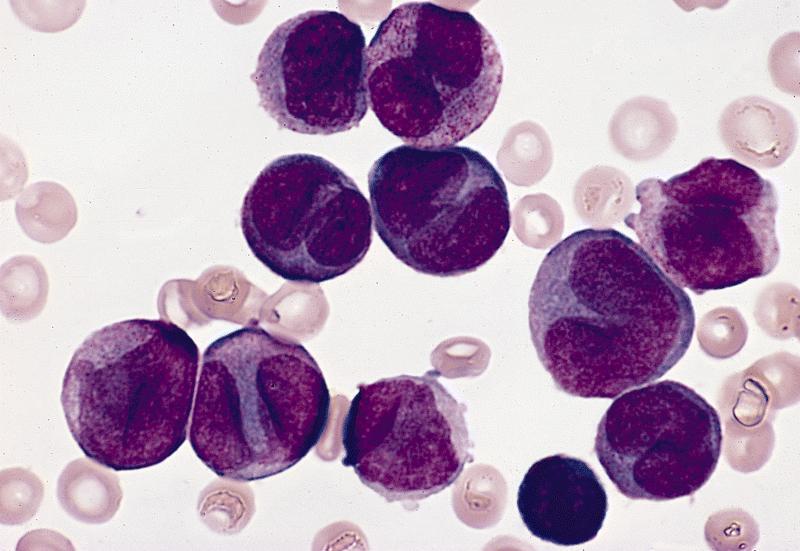
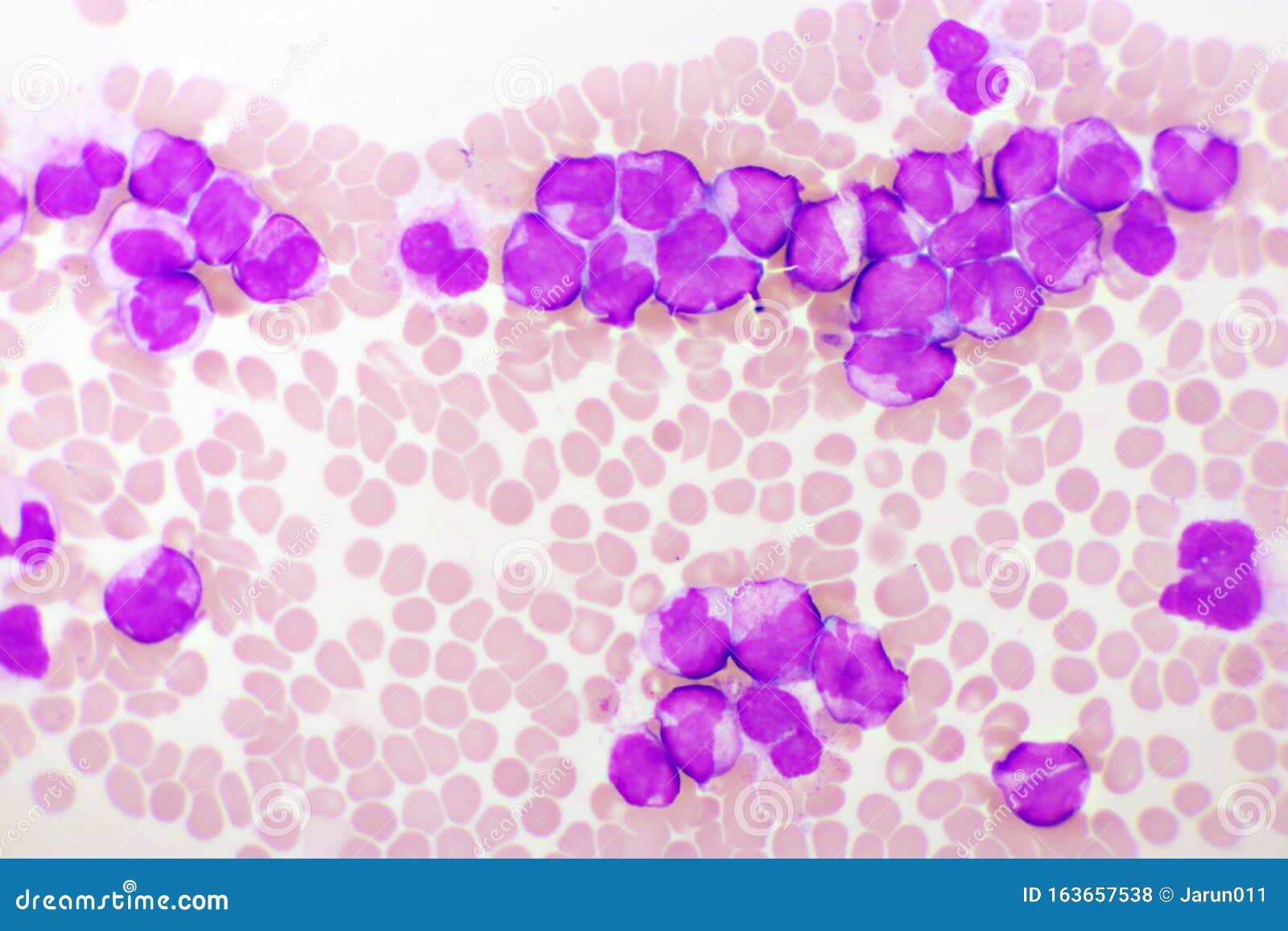
Paso 1: Morfología. La LPA se distingue por la presencia predominante de promielocitos anormales en la médula ósea y la sangre periférica. Estos promielocitos típicamente muestran una morfología anormal, con abundante citoplasma y gránulos azurófilos gruesos. Un ejemplo es la presencia de cuerpos de Auer múltiples ("faggot cells").
Paso 2: Inmunofenotipo. El inmunofenotipo de la LPA suele mostrar positividad para marcadores mieloides como CD13 y CD33, y típicamente es negativo para HLA-DR. Un ejemplo específico es la expresión fuerte de CD33 y la ausencia de HLA-DR, lo que ayuda a diferenciarla de otras LMA.
Must Read
Paso 3: Genética. La característica definitoria de la LPA es la translocación cromosómica t(15;17)(q22;q12), que fusiona el gen PML (promyelocytic leukemia) en el cromosoma 15 con el gen RARα en el cromosoma 17. Esta fusión crea el gen de fusión PML-RARα. Por ejemplo, la identificación de este gen por PCR o FISH confirma el diagnóstico de LPA.
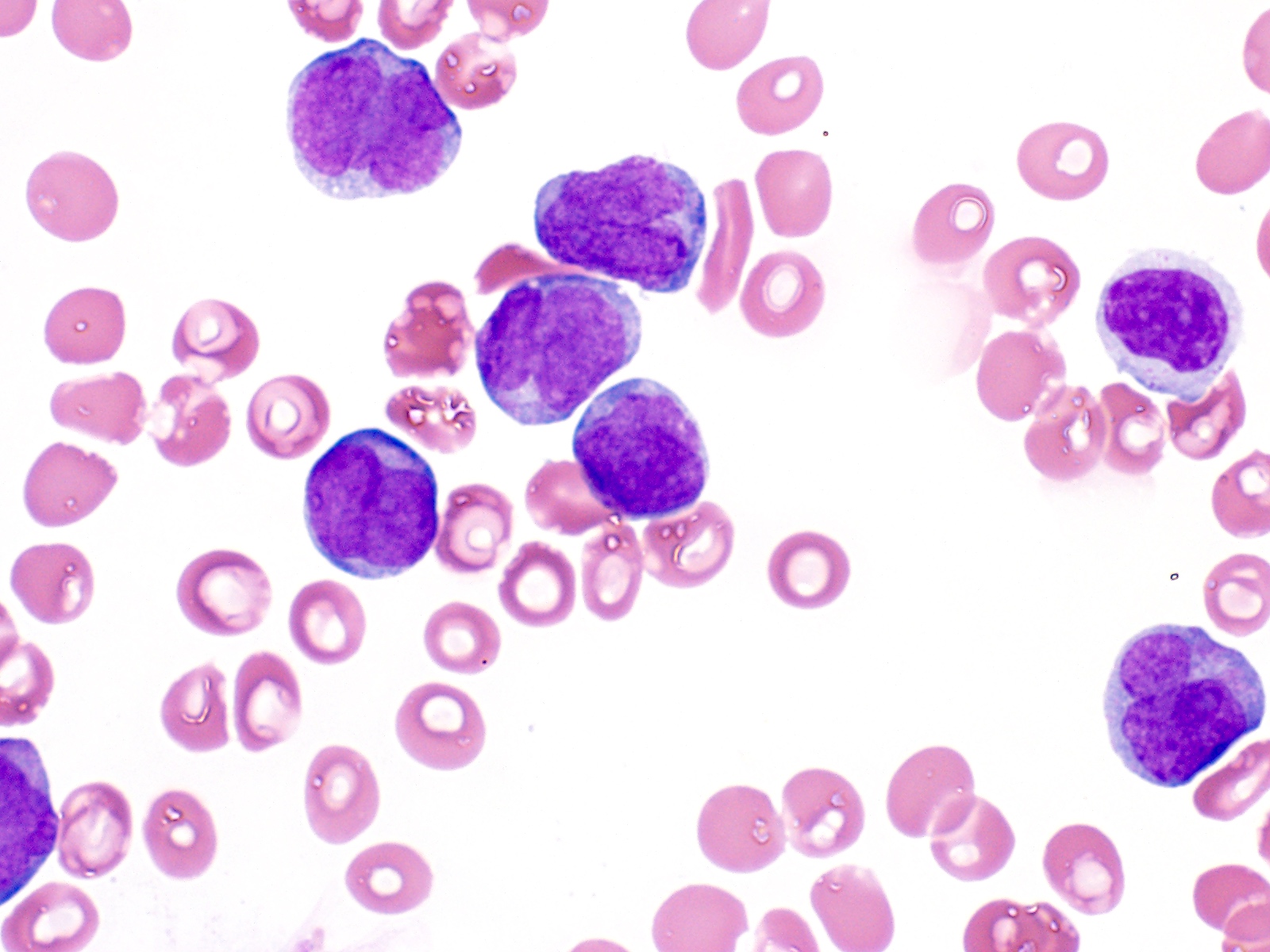
Paso 4: Diagnóstico. El diagnóstico de LPA se basa en la integración de la morfología, el inmunofenotipo y la genética. La detección del gen de fusión PML-RARα es crucial. Por ejemplo, si un paciente presenta promielocitos atípicos en la médula ósea y la PCR detecta PML-RARα, el diagnóstico de LPA está prácticamente confirmado.
Uso práctico 1: El diagnóstico preciso de LPA es crucial porque responde muy bien al tratamiento con ácido transretinoico (ATRA) y trióxido de arsénico (ATO). Uso práctico 2: Identificar rápidamente LPA permite iniciar el tratamiento temprano, reduciendo el riesgo de complicaciones como la coagulopatía intravascular diseminada (CID), una complicación frecuente y potencialmente fatal de esta leucemia.